Classical twin data comprise of phenotypic measurements on monozygotic (MZ) and dizygotic (DZ) twin pairs who were raised together. To derive estimates of behavioral genetic parameters (e.g., heritability) from such data, the ACDE model is most often used. In principle, the model provides estimates of the effects of additive genes (A), the shared environment (C), non-additive genes (D), and the unshared environment (E).
However, if only classical twin data are available, there is not enough information to estimate all four parameters, that is, the system of equations is underdetermined or underidentified. To enable parameters to be estimated, it is customary to fix either D or C to zero, leading to the ACE and ADE models which are identified. The problem with this approach is that if the influence of the omitted parameter is not actually zero, the estimates will be biased. Additional data on other types of family members, such as adoptees, would be needed for the full model but such data are usually not readily available.
Against this backdrop, Jöreskog (2021a) proposed that the full ACDE model can be estimated with only classical twin data. (A version of the ACDE model for categorical data was developed in Jöreskog [2021b], while Jöreskog [2021a] concerns only continuous data. I will discuss only the latter, but the same arguments apply to the categorical case.) This is a startling claim because the ACDE model has long been regarded as obviously impossible to estimate as there is simply not enough information in the twin variances and covariances for the full model (MZ and DZ variance-covariance matrices are sufficient statistics for the typical twin model, i.e., no other aspect of the sample data provides additional information on the parameter values). Nevertheless, Jöreskog claimed that it can be done, demonstrating it in several examples. Karl Jöreskog is not a behavioral geneticist but he is a highly influential statistician whose work on structural equation models has had a major influence on twin research. Therefore, even though his claims sounded implausible, they seemed worth investigating.
After studying Jöreskog’s model in detail I conclude that it does not deliver what it promises. It does generate a set of estimates for A, C, D, and E, but there is no reason to believe that they reflect the true population parameters. As nice as it would be to estimate the ACDE model with ordinary twin data, it just cannot be done.
This post has the following structure. I will start with a brief overview of twin models, describing some of the ways in which their parameters can be estimated. Then I will show how Jöreskog proposes to solve the ACDE identification problem, and where he goes wrong. I will end with a discussion of why I think twin models are useful despite their limitations, and why they have continuing relevance in the genomic era. The Appendix contains additional analyses related to the ACDE model.
[toc]
1. ACDE and its submodels
The ACDE model can be depicted as the following structural equation model (from Neale & Maes, 2004, p. 116):

A, C, D, and E latent causal variables. They stand for additive genetic effects, the shared environment, non-additive genetic effects, and the unshared environment, respectively. They have unit variances for convenience. There is a full set of four latent variables for each twin in a pair, as indicated by the subscripts. A and D can be thought of as the perfect additive and non-additive polygenic scores for the trait, while C and E can be interpreted analogously as “polyenvironmental” scores that capture all environmental differences between families (C) and within families (E). None of the four can be directly observed but assuming that the model is correct and identified, the influence of each on the phenotype can be estimated. a, c, d, and e are the free parameters to be estimated, representing the influences of the latent variables on 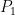

The free parameters a, c, d, and e can be interpreted like regression weights. For example, a one unit change in the latent variable C is expected to change an individual’s phenotypic score (

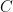




The graph above contains two models, one for MZ twins and the other for DZ twins. The two models are identical, explaining phenotypes with the same parameters, except for differences in correlations between the latent variables. MZ cotwins have perfect correlations for all genetic effects, while for DZ cotwins these correlations are smaller (0.50 or 0.25). The environmental correlations are 1 for C and 0 for E, and do not differ by zygosity. In MZ twins, all within-pair differences are due to E, while in DZ twins, within-pair differences can be due to A, D, or E. Other than the familial correlations, all the A, C, D, and E variables are uncorrelated with each other so that, for example, genetic and environmental components are not correlated. A, C, D, and E are assumed to be normally distributed.
The model-implied twin variances and covariances for the phenotype P, described in terms of a, c, d, and e, can be read off the graph using path-tracing rules:

The variances of
Apart from path-tracing, the variance and covariance expressions can be understood more intuitively as well. In particular, the amount of phenotypic variance explained by each effect is given by its square, so, with the assumption that the effects are uncorrelated, phenotypic variance equals the sum of all squared effects: 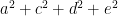


Following the approach in Jöreskog (2021a), the ACDE model can be written as a system of three equations:

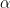
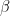
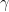
The first equation in (1.3) can be simplified with the help of the second equation:

As 
The most popular way to deal with the ACDE underidentification problem is to set the value of

Now there are three equations and three unknowns, so a unique solution is available. Through basic algebraic manipulations, we can express the three unknown parameters in terms of the known variance and covariances:

Substituting a sample variance and sample covariances for
Jöreskog develops his model in matrix form, so it is useful to do the same here. The ACE model can be written as the following matrix equation which is equivalent to (1.3) after the removal of the

The coefficient matrix is square and full rank, so a good way to solve for the unknowns is to multiply both sides with the inverse of the coefficient matrix, which gives the same ACE solutions as in (1.6):

So far, we have retained the original units of the phenotype, but if we want to calculate standardized effect sizes (or proportions of variance), we must take ratios of the unstandardized estimates. For example, heritability is:

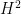
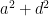



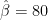


And therefore the heritability estimate is:

In the interests of generality, in this post I focus on twin models that produce unstandardized parameter estimates.[Note 2] However, standardized estimates can always be calculated as ratios of the unstandardized estimates, as above. In some examples I will use variance decompositions where the total variance equals 1, in which case the unstandardized and standardized models are the same model.
The ACE model fits most datasets fine, but if the MZ covariance is more than twice the DZ covariance, the ACE model

This is equivalent to the following matrix equation:

This coefficient matrix is invertible as well, so the equation can be solved by multiplying both sides with the inverse of the matrix:

At first blush, these estimators may seem mystifying. Why is it that, say, twice the MZ covariance minus four times the DZ covariance is expected to equal 
2. Jöreskog’s ACDE model
While the ACE and ADE models provide estimates of behavioral genetic parameters, they are based on the untested assumption that a particular parameter value is zero. This is a suboptimal approach with an obvious risk of bias, so Jöreskog’s claim that it is possible to estimate the full model without such uncertain assumptions is intriguing.
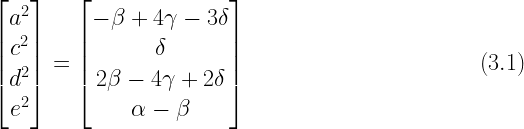
Jöreskog’s ACDE model can be represented by this matrix equation which is equivalent to the system of equations presented in (1.3) above:
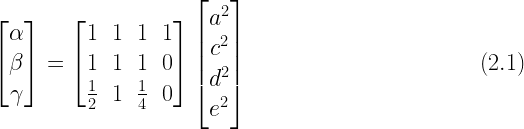
There are four unknowns (

A unique value can be calculated for 

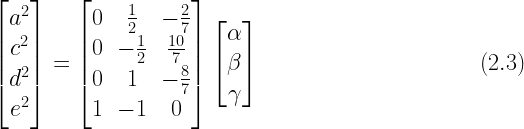
In this system, there is a one-to-one relationship between the unknown parameters and the known values

Standardized parameter estimates can be computed as ratios of the unstandardized estimates. For example, broad heritability is equal to:
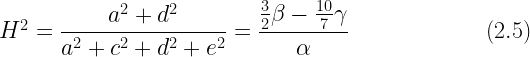
However, on the face of it, this solution is absurd. Why would this particular set of parameter estimates be anywhere close to the truth? For example, why would the additive genetic variance
To illustrate the problem, let’s say we have a phenotype with a variance of 1, and MZ and DZ covariances of 0.70 and 0.40, respectively (when the variance is 1, covariances are equivalent to correlations). The following variance decompositions are all consistent with the solution set given in (2.2):
| Model | $a^2$ | $c^2$ | $ d^2$ | $e^2$ | $ var$ | $cov_{MZ}$ | $ cov_{DZ}$ | $H^2$ |
|---|---|---|---|---|---|---|---|---|
| $ \text{I}$ | $ 0.60 $ | $ 0.10$ | $ 0.00$ | $ 0.30$ | $ 1.00$ | $ 0.70$ | $ 0.40$ | $ 0.60$ |
| $ \text{II}$ | $ 0.45$ | $ 0.15$ | $ 0.10$ | $ 0.30$ | $ 1.00$ | $ 0.70$ | $ 0.40$ | $ 0.55$ |
| $ \text{III}$ | $ 0.375$ | $ 0.175$ | $ 0.15$ | $ 0.30$ | $ 1.00$ | $ 0.70$ | $ 0.40$ | $ 0.525$ |
| $ \text{IV}$ | $ 0.15$ | $ 0.25$ | $ 0.30$ | $ 0.30$ | $ 1.00$ | $ 0.70$ | $ 0.40$ | $ 0.45$ |
| $ \text{V}$ | $ 0.075$ | $ 0.275$ | $ 0.35$ | $ 0.30$ | $ 1.00$ | $ 0.70$ | $ 0.40$ | $ 0.425$ |
| $ \text{Joreskog}$ | $ 0.24 $ | $ 0.22$ | $ 0.24$ | $ 0.30$ | $ 1.00$ | $ 0.70$ | $ 0.40$ | $ 0.48$ |
Innumerable different combinations of
The answer is simple. When the Moore-Penrose inverse is used to solve an underdetermined system with more unknowns than equations, it selects from among the infinite feasible solutions the one that minimizes the Euclidean norm, which is also known as the 
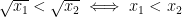






The solution is the vector whose squared entries have the smallest sum among all the vectors that satisfy equation (2.2). It is easy to see what kind of vectors fulfill this criterion best. Firstly, because squared quantities increase faster than linearly, it is better to have more rather than fewer non-zero entries in the vector whose distance from zero is being minimized. For example, it is better to have two parameters that each account for 40 units of variance than a single parameter that accounts for 80 units. This is because 
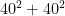
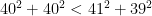
Therefore, of the infinitely many solutions that perfectly fit the underdetermined system of equations, Jöreskog's model selects the one that spreads the phenotypic variance as evenly as possible among all four parameters. The ideal ACDE solution under this criterion would be for each parameter to get one fourth of the variance, but because the solution must also be consistent with equation (2.2), the ideal can usually only be approximated. Jöreskog gives no theoretical reason for why there would be this tendency towards an even split between these four sources of variance, and it is difficult to come up with one. There may be applications where the Euclidean norm leads to a reasonable approximation in the absence of other information, but behavioral genetics is not one of those applications.
The biases of Jöreskog’s estimators are easy to investigate by plugging the model-implied values of the twin variance and covariances from (1.3) into the formulas in (2.4). The asymptotic expected values, ![E_\infty[\cdot]](https://s0.wp.com/latex.php?latex=E_%5Cinfty%5B%5Ccdot%5D&bg=ffffff&fg=000&s=0&c=20201002)
![\null\hfill (2.7)\\ E_\infty[\hat{a}^2] = \frac{1}{2}\beta - \frac{2}{7}\gamma \\\\= \frac{1}{2}(a^2 + c^2 + d^2) - \frac{2}{7}(\frac{1}{2}a^2 + c^2 + \frac{1}{4}d^2)\\\\ = \frac{5}{14}a^2 + \frac{3}{14}c^2 + \frac{3}{7}d^2 \\\\\\ E_\infty[\hat{c}^2] = -\frac{1}{2}\beta + \frac{10}{7}\gamma = -\frac{1}{2}(a^2 + c^2 + d^2) + \frac{10}{7}(\frac{1}{2}a^2 + c^2 + \frac{1}{4}d^2) \\\\ = \frac{3}{14}a^2 + \frac{13}{14}c^2 - \frac{1}{7}d^2 \\\\\\ E_\infty[\hat{d}^2] = \beta - \frac{8}{7}\gamma = (a^2 + c^2 + d^2) - \frac{8}{7}(\frac{1}{2}a^2 + c^2 + \frac{1}{4}d^2) \\\\ = \frac{3}{7}a^2 - \frac{1}{7}c^2 + \frac{5}{7}d^2\\\\\\ E_\infty[\hat{e}^2] = \alpha - \beta = a^2 + c^2 + d^2 + e^2 - (a^2 + c^2 + d^2) \\\\= e^2](https://s0.wp.com/latex.php?latex=%5Cnull%5Chfill+%282.7%29%5C%5C++++E_%5Cinfty%5B%5Chat%7Ba%7D%5E2%5D+%3D+%5Cfrac%7B1%7D%7B2%7D%5Cbeta+-+%5Cfrac%7B2%7D%7B7%7D%5Cgamma+%5C%5C%5C%5C%3D+%5Cfrac%7B1%7D%7B2%7D%28a%5E2+%2B+c%5E2+%2B+d%5E2%29+-+%5Cfrac%7B2%7D%7B7%7D%28%5Cfrac%7B1%7D%7B2%7Da%5E2+%2B+c%5E2+%2B+%5Cfrac%7B1%7D%7B4%7Dd%5E2%29%5C%5C%5C%5C+%3D+%5Cfrac%7B5%7D%7B14%7Da%5E2+%2B+%5Cfrac%7B3%7D%7B14%7Dc%5E2+%2B+%5Cfrac%7B3%7D%7B7%7Dd%5E2+%5C%5C%5C%5C%5C%5C++++E_%5Cinfty%5B%5Chat%7Bc%7D%5E2%5D+%3D+-%5Cfrac%7B1%7D%7B2%7D%5Cbeta+%2B+%5Cfrac%7B10%7D%7B7%7D%5Cgamma+%3D+-%5Cfrac%7B1%7D%7B2%7D%28a%5E2+%2B+c%5E2+%2B+d%5E2%29+%2B+%5Cfrac%7B10%7D%7B7%7D%28%5Cfrac%7B1%7D%7B2%7Da%5E2+%2B+c%5E2+%2B+%5Cfrac%7B1%7D%7B4%7Dd%5E2%29+%5C%5C%5C%5C+%3D+%5Cfrac%7B3%7D%7B14%7Da%5E2+%2B+%5Cfrac%7B13%7D%7B14%7Dc%5E2+-+%5Cfrac%7B1%7D%7B7%7Dd%5E2+%5C%5C%5C%5C%5C%5C++++E_%5Cinfty%5B%5Chat%7Bd%7D%5E2%5D+%3D+%5Cbeta+-+%5Cfrac%7B8%7D%7B7%7D%5Cgamma+%3D+%28a%5E2+%2B+c%5E2+%2B+d%5E2%29+-+%5Cfrac%7B8%7D%7B7%7D%28%5Cfrac%7B1%7D%7B2%7Da%5E2+%2B+c%5E2+%2B+%5Cfrac%7B1%7D%7B4%7Dd%5E2%29+%5C%5C%5C%5C+%3D+%5Cfrac%7B3%7D%7B7%7Da%5E2+-+%5Cfrac%7B1%7D%7B7%7Dc%5E2+%2B+%5Cfrac%7B5%7D%7B7%7Dd%5E2%5C%5C%5C%5C%5C%5C++++E_%5Cinfty%5B%5Chat%7Be%7D%5E2%5D+%3D+%5Calpha+-+%5Cbeta+%3D+a%5E2+%2B+c%5E2+%2B+d%5E2+%2B+e%5E2+-+%28a%5E2+%2B+c%5E2+%2B+d%5E2%29+%5C%5C%5C%5C%3D+e%5E2&bg=ffffff&fg=000&s=2&c=20201002)
Only the
To confirm that Jöreskog’s estimates reflect only the minimization described in (2.6), in the Appendix I show that his ACDE estimators can be derived using constrained optimization. Furthermore, a popular method for minimizing the
3. Euclidean norm versus parsimony
Many mathematical criteria other than the Euclidean norm could be used to come up with a solution to the underidentified ACDE model, leading to quite different sets of parameter estimates. For example, one could argue that parsimony should be favored, and in that case the ACE model which explains the data in the table in the previous section with three parameters (Model I) would obviously be preferred to the full ACDE model that requires four parameters to account for the same data. It could even be said that Jöreskog’s model reflects the principle of anti-parsimony, or Occam’s butterknife, with the most convoluted answer regarded as the best one. For example, even if the MZ covariance is exactly twice the DZ covariance, and the data are therefore consistent with the two-parameter AE model, Jöreskog’s approach will produce an ACDE solution where the variance is distributed approximately equally between the four parameters. Considering that many lines of evidence point to the AE model being the mechanism behind the typical complex human trait (Hill et al., 2008; Polderman et al., 2015), a widespread adoption of Jöreskog’s ACDE model would lead to a severely skewed understanding of genetic and environmental determinants of human variation.
An ACDE estimator that favors parsimony can be implemented with linear programming. The 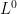
| $ a^2$ | $ c^2$ | $ d^2$ | $ e^2$ | |
|---|---|---|---|---|
| $ L^0$ solution 1 | 0.60 | 0.10 | 0.00 | 0.30 |
| $ L^0$ solution 2 | 0.00 | 0.30 | 0.40 | 0.30 |
The first solution is the ACE model, which at three parameters is more parsimonious than the ACDE model, but there is also a second, equally parsimonious solution, the CDE model. While the CDE model is algebraically feasible, it is very improbable–interactions without main effects are unusual in general, and non-additive genetic variance in the absence of additive genetic variance may well be impossible for a polygenic trait in any population that is not highly inbred.
Let’s assume that 
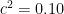


| $ a^2$ | $ c^2$ | $ d^2$ | $ e^2$ | |
|---|---|---|---|---|
| $ L^0$ solution 1 | 0.75 | 0.00 | 0.10 | 0.15 |
| $ L^0$ solution 2 | 0.00 | 0.25 | 0.60 | 0.15 | Joreskog | 0.31 | 0.15 | 0.39 | 0.15 |
The ADE and CDE models are now the parsimonious solutions. However, both are biased as the true model is ACDE. Jöreskog’s ACDE decomposition of these covariances, shown in the last row, is closer to the mark, but still quite biased. Even when ACDE is the true model, Jöreskog’s estimates will be satisfactory only by chance.
As should be obvious from these examples, there is no reason to believe that the Euclidean norm, parsimony, or any other mathematically convenient criterion would produce ACDE estimates that are anywhere close to the true population values. If the full model is to be estimated, there are no substitutes for acquiring additional data that are informative about 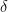
4. Discussion
Jöreskog (2021a) proposed to estimate the ACDE model by using the Moore-Penrose inverse. This enabled him to come up with estimates for all four parameters (
ACE is the most commonly used twin model. It assumes that genetic effects are purely additive, but if this is not the case, estimates will be biased. Nevertheless, I would argue that the bias is not too serious in typical settings. The asymptotically expected amount of bias can be found by plugging an ACDE decomposition in the
![E_\infty[\hat{a}^2] = 2(cov_{MZ} - cov_{DZ}) \\\\= 2(a^2 + c^2 + d^2 - [\frac{1}{2}a^2 - c^2 - \frac{1}{4}d^2]) \hfill (4.1) \\\\= a^2 + \frac{3}{2}d^2 = a^2 + d^2 + \frac{1}{2}d^2\\\\\\ E_\infty[\hat{c}^2] = 2cov_{DZ} - cov_{MZ} \\\\= 2(\frac{1}{2}a^2 + c^2 + \frac{1}{4}d^2) - (a^2 + c^2 + d^2) \hfill (4.2)\\\\= c^2 - \frac{1}{2}d^2](https://s0.wp.com/latex.php?latex=E_%5Cinfty%5B%5Chat%7Ba%7D%5E2%5D+%3D+2%28cov_%7BMZ%7D+-+cov_%7BDZ%7D%29+%5C%5C%5C%5C%3D+2%28a%5E2+%2B+c%5E2+%2B+d%5E2+-+%5B%5Cfrac%7B1%7D%7B2%7Da%5E2+-+c%5E2+-+%5Cfrac%7B1%7D%7B4%7Dd%5E2%5D%29++%5Chfill+%284.1%29+%5C%5C%5C%5C%3D+a%5E2+%2B+%5Cfrac%7B3%7D%7B2%7Dd%5E2+%3D+a%5E2+%2B+d%5E2+%2B+%5Cfrac%7B1%7D%7B2%7Dd%5E2%5C%5C%5C%5C%5C%5C++E_%5Cinfty%5B%5Chat%7Bc%7D%5E2%5D+%3D+2cov_%7BDZ%7D+-+cov_%7BMZ%7D+%5C%5C%5C%5C%3D+2%28%5Cfrac%7B1%7D%7B2%7Da%5E2+%2B+c%5E2+%2B+%5Cfrac%7B1%7D%7B4%7Dd%5E2%29+-+%28a%5E2+%2B+c%5E2+%2B+d%5E2%29+%5Chfill+%284.2%29%5C%5C%5C%5C%3D+c%5E2+-+%5Cfrac%7B1%7D%7B2%7Dd%5E2+&bg=ffffff&fg=000&s=2&c=20201002)
When non-additive genetic effects are present, the ACE estimate of genetic variance in (4.1) does not approximate the additive genetic variance, but is instead an inflated estimate of the total genetic variance (or the unstandardized broad heritability). The expected inflation factor is equal to 
Another thing to consider is that human mating is not random with respect to many socially valued phenotypes studied by behavioral geneticists. This means that bias in ACE estimates due to a neglect of non-additive heredity is often counterbalanced by an opposite bias due to the unmodeled effects of assortative mating. Moving from random mating to (positive) assortative mating increases phenotypic variance in the same way in MZ and DZ twins.[Note 4] It does not change the MZ genetic correlation (which is always perfect), but it increases the DZ additive genetic covariance in relation to the MZ additive genetic covariance, so that the former will now be more than half the size of the latter, on average. If p is the phenotypic correlation between parents and mate selection is based directly on phenotypic similarity, the expected covariance of DZ twin offspring under the ACDE model is (see Lynch & Walsh, 1998, p. 158):
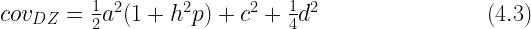
where
![E_\infty[\hat{a}^2] = 2(cov_{MZ} - cov_{DZ}) \\\\= 2(a^2 + c^2 + d^2 - \frac{1}{2}a^2[1 + h^2p] - c^2 - \frac{1}{4}d^2) \hfill (4.4)\\\\ = a^2 - a^2h^2p + \frac{3}{2}d^2 = a^2 + d^2 + \frac{1}{2}d^2 - a^2h^2p\\\\\\ E_\infty[\hat{c}^2] = 2cov_{DZ} - cov_{MZ} \\\\= 2(\frac{1}{2}a^2[1 + h^2p]) + c^2 + \frac{1}{4}d^2) - a^2 - c^2 - d^2 \hfill (4.5)\\\\ = c^2 - \frac{1}{2}d^2 + a^2h^2p](https://s0.wp.com/latex.php?latex=E_%5Cinfty%5B%5Chat%7Ba%7D%5E2%5D+%3D+2%28cov_%7BMZ%7D+-+cov_%7BDZ%7D%29+%5C%5C%5C%5C%3D+2%28a%5E2+%2B+c%5E2+%2B+d%5E2+-+%5Cfrac%7B1%7D%7B2%7Da%5E2%5B1+%2B+h%5E2p%5D+-+c%5E2+-+%5Cfrac%7B1%7D%7B4%7Dd%5E2%29+%5Chfill+%284.4%29%5C%5C%5C%5C++%3D+a%5E2+-+a%5E2h%5E2p+%2B+%5Cfrac%7B3%7D%7B2%7Dd%5E2+%3D+a%5E2+%2B+d%5E2+%2B+%5Cfrac%7B1%7D%7B2%7Dd%5E2+-+a%5E2h%5E2p%5C%5C%5C%5C%5C%5C++E_%5Cinfty%5B%5Chat%7Bc%7D%5E2%5D+%3D+2cov_%7BDZ%7D+-+cov_%7BMZ%7D+%5C%5C%5C%5C%3D+2%28%5Cfrac%7B1%7D%7B2%7Da%5E2%5B1+%2B+h%5E2p%5D%29+%2B+c%5E2+%2B+%5Cfrac%7B1%7D%7B4%7Dd%5E2%29+-+a%5E2+-+c%5E2+-+d%5E2+%5Chfill+%284.5%29%5C%5C%5C%5C++%3D+c%5E2+-+%5Cfrac%7B1%7D%7B2%7Dd%5E2+%2B+a%5E2h%5E2p+&bg=ffffff&fg=000&s=2&c=20201002)
The genetic variance estimate in (4.4) approximates the total genetic variance and is still affected by the upwardly biasing 





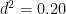



While ACE is not the model that generated the data, the heritability estimate nevertheless equals broad heritability, and the shared environment estimate is right on the mark. In real data, the result would probably not be as perfect, but the fact that biases in the ACE model frequently go in opposite directions does tend to blunt the distortionary effects of model violations. This kind of mutual canceling may also explain why the DZ covariance is often not more than half the MZ covariance for phenotypes that are known to be subject to positive assortative mating.
Contrary to the notion that progress is genomics would swiftly make twin studies and kinship-based behavioral genetics in general obsolete, twin and family studies remain essential for understanding human genetics. While these traditional methods cannot estimate the genetic values of individuals, they nevertheless have certain advantages over genomic methods.
A particular problem with genomic methods is population stratification which means that the apparent effect of genetic locus G on phenotype P may not be causal (

Another advantage of twin and family methods is that they can estimate the total genetic variance, while genomic methods have so far tended to be limited to the effects of genetic variants that are common (e.g., with a population frequency greater than 1 percent). Whole-genome data and genomic methods for analyzing related individuals make the bridging of this gap possible, and the expectation is that genomic estimates of heritability will converge to those from twin and family studies. Until either all of family-based heritability for a given trait has been explained genomically, or any discrepancies between family and genomic estimates have been convincingly resolved otherwise, there is no full explanation of familial phenotypic resemblance. Triangulation with different methods subject to different weaknesses is a traditional approach to debiasing estimates in behavioral genetics, and it comes to good use when genomic and kinship-based estimates are harmonized.
Notes
1. Non-additive genetic effects are referred to with the symbol D or d, but it should be noted that in the classical twin design this source of influence includes both dominance and epistasis, not just dominance. This is so because the MZ correlation for all genetic effects, additive and non-additive, is 1, while the DZ correlation is 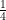
This symmetry breaks down if there is higher-order epistasis which means that three or more alleles interact with each other. The MZ correlation is 1 for all higher-order effects, too, while the DZ correlation for each effect is 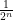
However, both empirical and theoretical evidence indicates that non-additive genetic variance is unlikely to pose serious problems to twin modeling. Firstly, if non-additive variance and higher-order interactions in particular were major determinants of phenotypic variance, MZ twin pairs would commonly be much more than twice as similar as DZ twins. In their meta-analysis of almost 18,000 traits in 14.5 million twin pairs, Polderman et al. (2015) found that for 69 percent of traits, the observed twin correlations were consistent with a model where twin resemblance was solely due to additive genetic variation, i.e., MZ twins were twice as similar as DZ twins. For most of the traits for which this model was inadequate the MZ pairs were less than twice as similar as DZ pairs, which is also inconsistent with non-additive genetic variance being a major determinant of phenotypic differences. Thus only a small proportion of traits show evidence of important gene interactions, contradicting the notion that complex interactions, rather than genetic and environmental main effects, are essential to explaining human differences.
Non-additive genetic variance has also been investigated with genomic methods. These methods fail to support the idea that dominance or epistasis would be important causes of phenotypic differences in humans (Pazokitoroudi et al., 2021; Hivert et al., 2021a). Consistently with the twin data, genomic methods suggest that the non-additive genetic contribution could be zero for the typical trait. In the behavioral domain, Okbay et al. (2022) notably found no evidence for dominance effects on educational attainment in a well-powered genome-wide association sample of 2.5 million people. (The existence of recessive mental retardation syndromes and inbreeding depression indicate that dominance effects on educational attainment are non-zero, but such effects cannot be observed in GWASs. Given the rarity of these conditions, at least in developed countries, the proportion of population variance that they contribute must be very small.)
Several theoretical and mathematical explanations of the observation that genetic variance is overwhelmingly additive have been developed. The basic insight of these arguments is that even if there is non-additive gene action in individuals, it does not necessarily translate into non-additive genetic differences between individuals. Rather, much or all of the non-additive influence will be reflected in the additive (or average) effects of loci. Hill et al. (2008) argued that the allele frequency distributions of human traits are usually U-shaped, with most frequencies close to 0 or 1, causing the average effects to account for essentially all of the variance even when there is dominant or epistatic gene action.
Mäki-Tanila & Hill (2014) emphasize that all “the epistatic terms involving a particular locus appear in its average effect”, which means that epistasis as such does not lead to large epistatic variance. They report that both uniform and U-shaped allele frequency distributions result in mostly additive variance, while frequencies close to 50 percent yield the most epistatic variance, although with a large number of loci epistatic variance will be low for all frequencies. For example, the frequency distribution of the nearly 4,000 genome-wide significant effect alleles in the educational attainment GWAS by Okbay et al. (2022) is roughly uniform (and would be more U-shaped if rare alleles were observed):
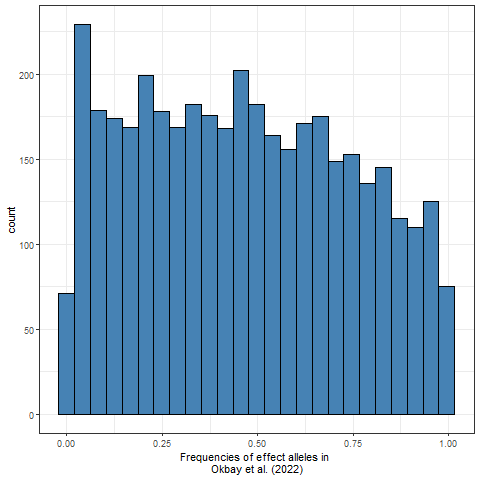
A predominance of allele frequencies close to 50 percent would be expected only in artificial selection experiments, which also helps explain why high epistatic variances have been reported in some studies on model organisms. Mäki-Tanila & Hill (2014) also point out that “epistatic variance is almost entirely contributed by the two-locus epistatic variance”, indicating that higher-order interactions are not expected to contribute much. This suggests that the assumption in twin modeling that the DZ correlation for non-additive genetic effects is always
These considerations indicate that non-additive genetic variance will not be a large source of phenotypic variance for many, or any, complex human traits. Nevertheless, it may often be a small source of variance, and its omission from twin models may cause some bias.
Unmodeled gene-environment interaction is another potential source of bias. It is similar to non-additive genetic variance in that it is often postulated to be important but in practice can rarely be observed with any reliability. It also seems that similar mathematical arguments that point to the rarity of dominance and epistatic variance could be made regarding gene-environment interaction variance.
2. If unstandardized effects are of no interest, the ACE model can also be estimated by using MZ and DZ correlations. The fact that there are multiple estimates of phenotypic variance is ignored, which means that the resulting estimates concern only proportions of variance, not the absolute amounts of variance explained by each parameter. The MZ and DZ phenotypic variances are therefore allowed, in principle, to differ by any amount, whereas unstandardized ACE models must either constrain MZ and DZ variances to equality, or to explicitly include unequal variances in the model specification. The standardized ACE model is:

The estimators are:
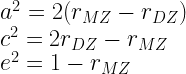


3. Jöreskog describes (1.6), (1.12), and (2.4) as maximum likelihood estimators, but as far as I can see, maximum likelihood is incidental to this approach, used only to estimate 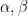
4. Under random mating the effects of genetic loci are uncorrelated (assuming that other mechanisms, e.g., natural selection, do not cause correlations), so that the total variance of the effects of all loci equals the sum of the variances of the effects. In contrast, under assortative mating the total variance equals the sum of the variances of the effects plus their covariances. This comes from a basic relationship between variances and covariances:

The covariances between effects for each locus are zero under random mating, while under positive assortative mating the covariances are positive, leading to increased variance. If there is disassortative mating (“opposites attract”), the covariance terms will be negative, and the genetic variance will be smaller than under random mating. However, it seems dubious whether disassortative mating exists in humans, other than in the trivial case of the perfect negative correlation between the parents’ sexes.
If (positive) assortative mating stays at the same level for several generations, additive genetic variance will reach a constant equilibrium value 

This formula can be used to estimate how much lower the additive genetic variance of the trait would be without assortative mating.
5. Method-of-moments ACE and ADE estimators that accommodate assortative mating can be derived from (1.6), (1.12), and (4.3).
The ACE estimators are:
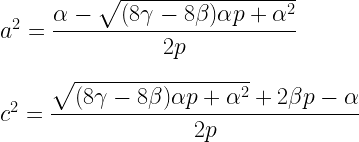
The ADE estimators are:
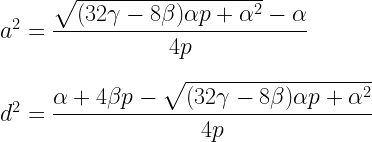
The assumption in both sets of estimators is that assortative mating has been going on for several generations while p, the parental phenotypic correlation has remained constant, and therefore
References
Hill, W. G., Goddard, M. E., & Visscher, P. M. (2008). Data and theory point to mainly additive genetic variance for complex traits. PLoS Genet., 4, e1000008.
Hill, W. G., & Mäki-Tanila, A. (2015). Expected influence of linkage disequilibrium on genetic variance caused by dominance and epistasis on quantitative traits. J. Anim. Breed. Genet., 132, 176–186.
Hivert, V., et al. (2021a). Estimation of non-additive genetic variance in human complex traits from a large sample of unrelated individuals. Am. J. Hum. Genet., 108, 786–798.
Hivert V., Wray N. R., & Visscher, P. M. (2021b). Gene action, genetic variation, and GWAS: A user-friendly web tool. PLoS Genet 17(5): e1009548.
Jöreskog, K. G. (2021a). Classical models for twin data. Structural Equation Modeling: A Multidisciplinary Journal, 28, 121–126.
Jöreskog, K. G. (2021b). Classical Models for Twin Data: The Case of Categorical Data. Structural Equation Modeling: A Multidisciplinary Journal, 28, 859–862.
Lynch, M., & Walsh, B. (1998). Genetics and Analysis of Quantitative Traits. Sunderland, MA: Sinauer Associates.
Mäki-Tanila, A., & Hill, W. G. (2014). Influence of gene interaction on complex trait variation with multilocus models. Genetics, 198, 355–367.
Neale, M.C., & Maes, H.H.M. (2004). Methodology for genetic studies of twins and families. Dordrecht, NL: Kluwer Academic Publishers.
Okbay, A., et al. (2022). Polygenic prediction of educational attainment within and between families from genome-wide association analyses in 3 million individuals. Nature Genetics, 54, 437–449.
Pazokitoroudi, A., et al. (2021). Quantifying the contribution of dominance deviation effects to complex trait variation in biobank-scale data. Am. J. Hum. Genet., 108, 799–808.
Polderman, T. J. C., et al. (2015). Meta-analysis of the heritability of human traits based on fifty years of twin studies. Nature Genetics, 47, 702–709.
Appendix
Lagrange multipliers
Jöreskog’s model can also be estimated with the method of Lagrange multipliers. This method allows for the minimization of an objective function that is subject to equality constraints.
With $ a^2, c^2, d^2$, and $ e^2$ denoted by $ A, C, D$, and $ E$ for convenience, the objective function to be minimized is:
$ f = A^2 + C^2 + D^2 + E^2$
Equation (2.2) provides three constraints (the fourth is the identity $ d^2 = d^2$ which is of no use here) that are arranged so that all non-zero terms are on the same side:

The Lagrangian function is:
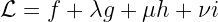
where the three Greek letters are the unknown Lagrange multipliers.
The partial derivatives of $ \mathcal{L}$ with respect to $ A, C, D$, and $ E$ are:

The four partial derivatives, when equated to 0, and the three constraints form a system of seven linearly independent equations in seven unknowns:

This system can be solved by multiplying both sides with the inverse of the coefficient matrix:
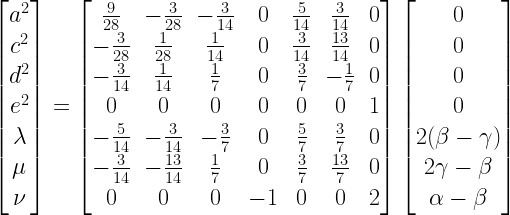
The solution is:

These values of $ a^2, c^2, d^2$, and $ e^2$ match exactly with the pseudoinverse solution presented in equation (2.4), and given that $ f$ is a strictly convex function, the values are where $ f$ reaches its minimum. This confirms that Jöreskog’s estimates are equivalent to
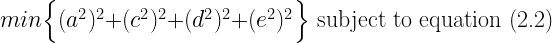
which, alas, provides no basis for estimating behavioral genetic parameters.
Ridge regression
Ridge regression minimizes the $ L^2$ norm, and can be used for underdetermined systems, so it should be equivalent to Jöreskog’s method. The estimator is:
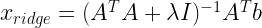
Applying it to equation (2.1), we get:

This results in a rather complex expression:

However, 

As expected, the answer matches the pseudoinverse solution in equation (2.4).
Ridge regression is typically used to mitigate the effects of multicollinearity in regression models with many highly correlated independent variables. It reduces the variance of the estimates in exchange for some bias. However, while ridge regression can be used to solve the underdetermined ACDE model, it is not a reasonable use case for it. The results are so biased that they are worthless.
Sparse solutions to the ACDE model
The $ L^0$ “norm” denotes the number of non-zero entries of a vector. Minimizing the $ L^0$ “norm” results in a parsimonious, or sparse, solution where there are as many zero entries as feasible. A sparse solution to the ACDE model is one where the parameter estimate vector $ \begin{bmatrix} a^2 & c^2 & d^2 & e^2 \end{bmatrix}^T$ contains as many zero entries as possible given the constraints stated in equation (2.2).
The $ L^0$ minimization problem can be solved iteratively with mixed integer linear programming. $ a^2, c^2, d^2$, and $ e^2$ are denoted $ A, C, D$, and $ E$ for convenience and to emphasize that for the optimizer they are just numbers, rather than squares of anything (polynomials involved in linear programming can be at most degree 1). The objective function to be minimized is the number of non-zero entries in $ \{ A, C, D, E \}$. The constraints describing the set of all feasible solutions are given by equation (2.2), with phenotypic variance and MZ and DZ covariances fixed to some observed values.
In the program, there is a binary count variable accompanying each continuous variable A, C, D, and E, and additional constraints are set up so that each binary variable gets a value of 0 when its associated continuous variable is zero and a value of 1 when the associated continuous variable is non-zero (this is a variant of the Big M method). The optimizer then finds solutions to the constraints that minimize the sum of the binary count variables. I used the R package lpSolve to do the optimization:
#install.packages("lpSolve")
#install.packages("lpSolveAPI")
library(lpSolve)
library(lpSolveAPI)
# twin variance and covariances
varMZDZ <- 225
covMZ <- 125
covDZ <- 38
# auxiliary constant to prevent small (co)variances from messing up the counting constraints
if(varMZDZ < 3)
aux <- 0.99 else
aux <- 0
# set the number of variables
model <- make.lp(0, 8)
# define the objective function; the 1s signify equal weights
set.objfn(model, c(0, 0, 0, 0, 1, 1, 1, 1))
# counter variables are binary
set.type(model, c(5, 6, 7, 8), "binary")
# set constraints to define the feasible solution set
add.constraint(model, c(1, 0, 1.5, 0, 0, 0, 0, 0), "=", 2*(covMZ - covDZ))
add.constraint(model, c(0, 1, -0.5, 0, 0, 0, 0, 0), "=", 2*covDZ - covMZ)
add.constraint(model, c(0, 0, 0, 1, 0, 0, 0, 0), "=", varMZDZ - covMZ)
# set constraints to count nonzero parameters
add.constraint(model, c(-1, 0, 0, 0, 1, 0, 0, 0), "<=", aux)
add.constraint(model, c(1, 0, 0, 0, -1*(covMZ), 0, 0, 0), "<=", 0)
add.constraint(model, c(0, -1, 0, 0, 0, 1, 0, 0), "<=", aux)
add.constraint(model, c(0, 1, 0, 0, 0, -1*(covMZ), 0, 0), "<=", 0)
add.constraint(model, c(0, 0, -1, 0, 0, 0, 1, 0), "<=", aux)
add.constraint(model, c(0, 0, 1, 0, 0, 0, -1*(covMZ), 0), "<=", 0)
# set the upper and lower bounds
set.bounds(model, lower=c(0, 0, 0, 0, 0, 0, 0, 0), upper=c(covMZ, covMZ, covMZ, varMZDZ - covMZ, 1, 1, 1, 1))
# compute the optimized model
solve(model)
solution_number <- 1
# get the minimized value of the objective function
objective <- get.objective(model)
# lpSolve does not automatically report multiple solutions at the same level of sparsity, so the program will loop until all maximally
# sparse solutions have been found, with a new constraint equivalent to the previous solution added after each loop
while (objective == get.objective(model) & objective<10) {
cat("\n\nOptimal solution #", solution_number)
cat("\nNonzero parameters:", get.objective(model))
cat("\na^2: ", get.variables(model)[1])
cat("\nc^2: ", get.variables(model)[2])
cat("\nd^2: ", get.variables(model)[3])
cat("\ne^2: ", get.variables(model)[4])
add.constraint(model, c(0, 0, 0, 0, get.variables(model)[5:7], 0), "<=", sum(get.variables(model)[5:7])-1)
solve(model)
solution_number <- solution_number + 1
}
At least one of $ \{ A, C, D \}$ can always be set to zero while satisfying the constraints–that is the logic behind the practice of fixing D or C to zero when estimating ACE and ADE models. The optimizer will therefore always find at least one solution that is more sparse than the full ACDE model.
Discover more from Human Varieties
Subscribe to get the latest posts sent to your email.
Leave a Reply

© 2026 Human Varieties
Theme by Anders Noren — Up ↑
Hi to whoever is writing this (sorry for necroposting). I was originally going to comment this question in another post more related to it but the comments were a lot and I didn’t want to mess up the structure more. I was redirected elsewhere by Meng, because he’s more into psychometric stuff and this question is more about the topic of heredity.
What do you make of the differences in heritabilities between different methods (like Young’s RDR)? Do you plan on making any posts about why they yield deflated estimates? I know this topic has been discussed a little on social media but I haven’t seen anyone go into detail about these methods